

NextGENe®LR
NextGENeLR Software is an effective and easy-to-use resource for the analysis of long read sequencing data such as data from Pacific Biosciences RS, RSII, Sequel, and Sequel II systems, as well as Oxford Nanopore MinION.
NextGENeLR can be used for structural variation detection, STR expansion analysis, and whole genome mitochondrial DNA analysis, including low frequency SNV and indel detection, structural variation detection, and mitochondrial haplotyping, with support for mixed samples.
Analyses can be set up using an intuitive interface which facilitates batch processing of a number of samples. Recent enhancements to instruments such as PACBIO® and Oxford Nanopore Technologies have improved accuracy to as much as 98%.
Structural Variation Detection
NextGENeLR provides fast and accurate alignments of long sequencing reads and enables accurate detection of structural variants.
NextGENeLR uses a specialized long read alignment algorithm. This alignment algorithm works by first indexing the reference sequence, compressing homopolymer regions, which allows homopolymer length differences to be tolerated during alignment. A separate index table is saved for each chromosome. Reads are also similarly indexed.
Each chromosome table is then searched for matches. The region with the most and longest matches is selected and a local alignment is performed. The long reads can be split to properly align reads containing structural variants. For each read the alignment positions for each section are compared to the reference. Any differences between the reads and reference are evaluated to determine the structural variant type, as shown in Figure 1 below.
If sections of a read align to different chromosomes than a translocation is reported. If sections of a read align in different directions, then an inversion is reported. Similar breakpoints, within 5bp, are merged.
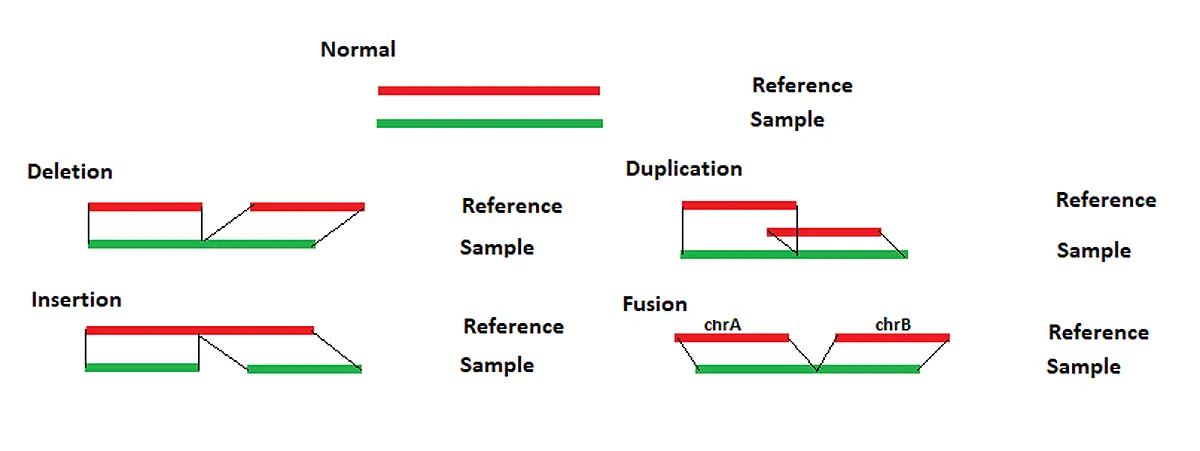
Figure 1: NextGENeLR makes structural variant calls by comparing the alignment positions for the sample and the reference. The patterns above illustrate how the structural variant type is reported.
Once analysis is completed the generated BAM file can be viewed using NextGENeLR’s Alignment Viewer. The Structural Variation Table is shown by default, with the Pileup Viewer above the table.
The Structural Variation Table lists the detected structural variants. For each variant, a type is provided as well as the number of reads supporting the structural variant, the breakpoint positions, the frequency of the variant out of the total reads aligned at each breakpoint position, and the distance in bp for the sample and the reference.
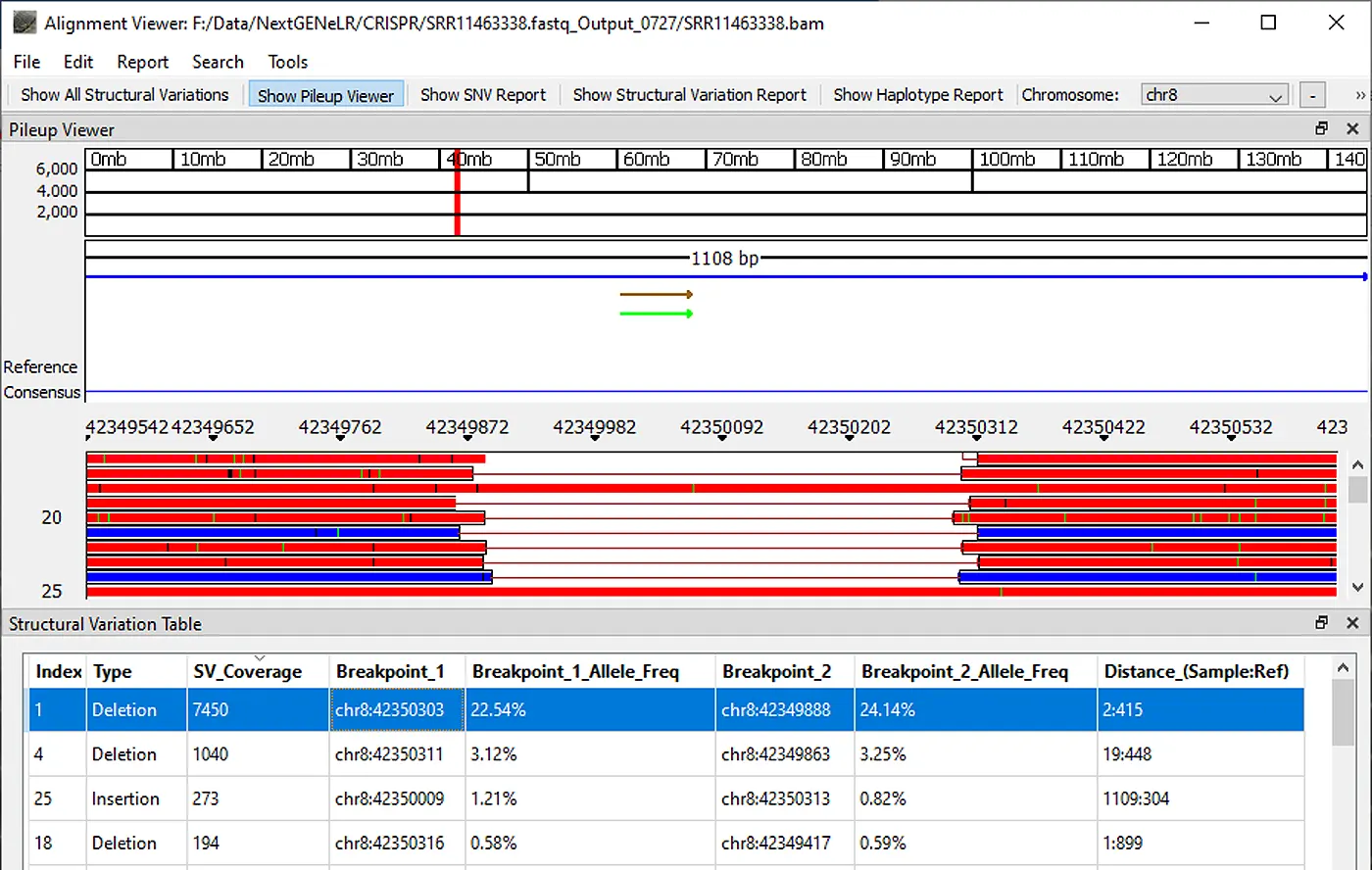
Figure 2: NextGENeLR Alignment Viewer showing the Structural Variation Table. Highlighted is a 413bp deletion.
Whole Genome Mitochondrial DNA Analysis
NextGENeLR software can be used for the analysis of whole genome mitochondrial DNA sequencing using long read data such as data from Pacific BioSciences systems. Pacific Biosciences SMRT (single molecule, real-time) sequencing, produces reads of dozens of kilobases in length, enabling the sequencing of up to the entire mitochondrial genome in one pass.
Whole genome mitochondrial DNA sequencing can be analyzed using NextGENeLR software for:
- Detection of structural variants
- Haplotyping, including mixed samples
- Detection of SNVs and indels, including low frequency variants
In conjunction with long read lengths of dozens of kilobases, NextGENeLR provides a powerful solution for the detection of structural variants. NextGENeLR software allows the accurate haplotyping of long read data by implementing accurate alignment and comparing aligned reads to reference haplotypes. Haplotypes can also be discerned from mixture samples, with detection as low as 0.1% frequency. NextGENeLR software can detect SNVs and small indels at frequencies as low as 2%.
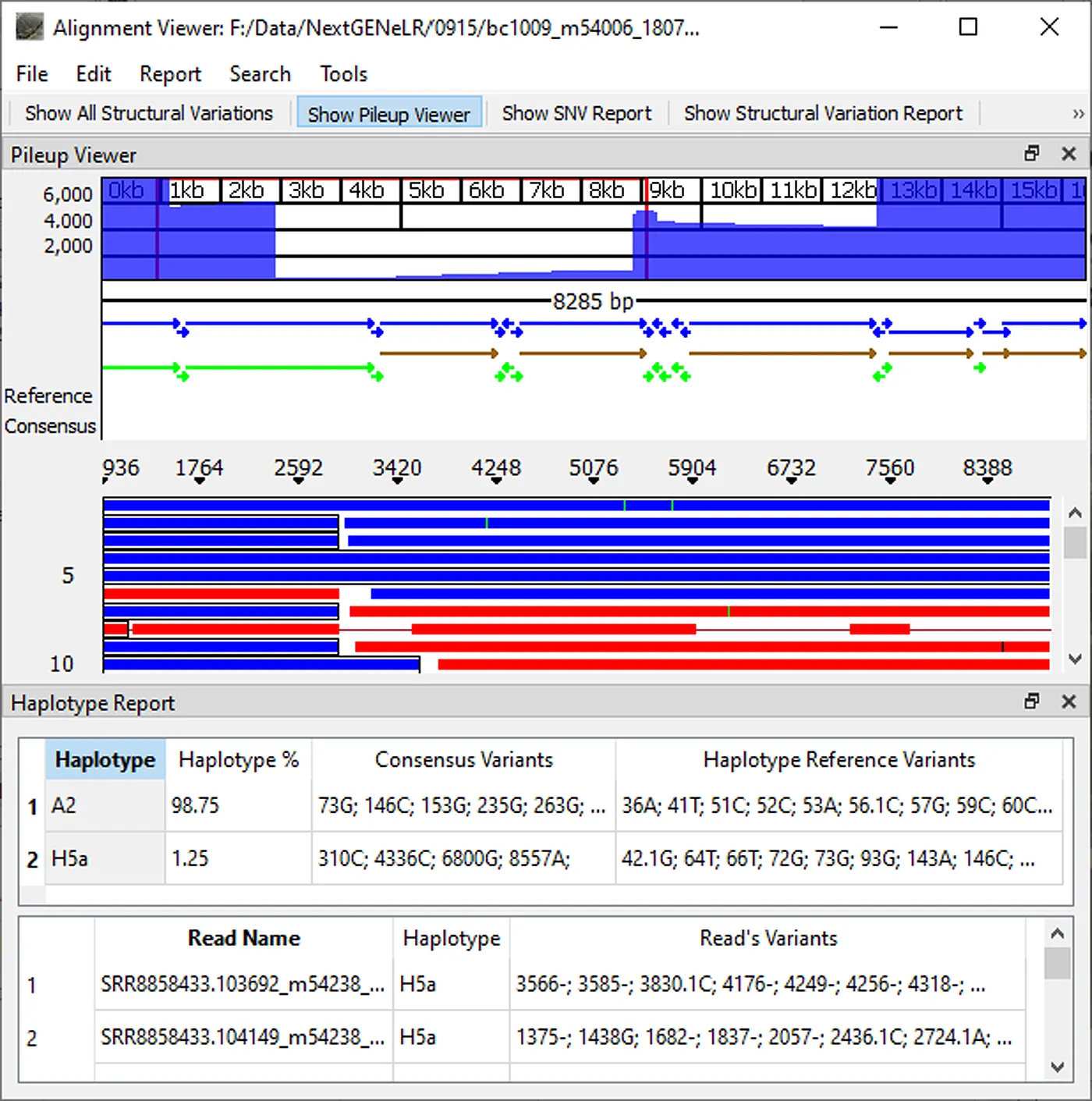
Figure 3: NextGENeLR Alignment Viewer showing the Structural Variation Table. Highlighted is a 413bp deletion.
STR Expansion Analysis
NextGENe®LR software can be used for STR Expansion analysis from long read sequencing data such as< data from Pacific BioSciences systems. This provides an accurate reporting of STR lengths to identify STR loci with lengths associated with disease.
NextGENeLR aligns the long sequence reads to the reference sequence using a specialized alignment algorithm. Alignment results are saved to a BAM file which can be loaded in the built-in Alignment Viewer for visualization and reporting.
When the STR Expansion Report is opened, the STR regions to be evaluated are defined. A default list of STR regions associated with disease is provided. This list of regions can be modified by removing regions, adding regions, editing regions, or uploading a text file containing a list of regions to be added. For the STR regions of interest, the number of reads with each repeat length is counted.
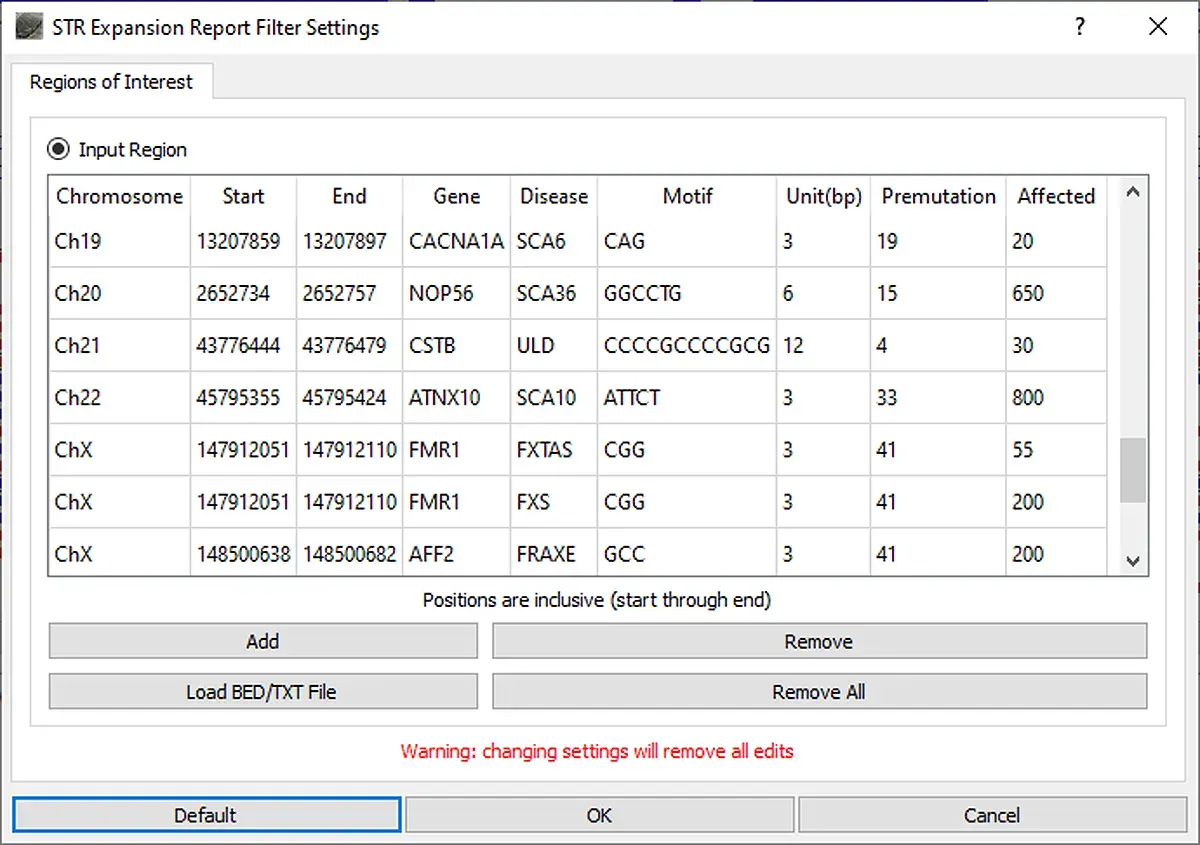
Figure 4: STR Expansion Regions of Interest can be defined. The default list can be used as is, regions can be removed and/or added, or a custom list of regions can be loaded.
Repeat lengths for all included regions of interest are displayed in the graphical STR Expansion Report. The normal range for the repeat length is displayed with green. Gray denotes the premutation range and red indicates the repeat length associated with disease. Blue boxes are placed at the repeat length(s) detected for the sample. The top number for each blue box indicates the repeat length and the bottom number indicates the number of reads with this length.
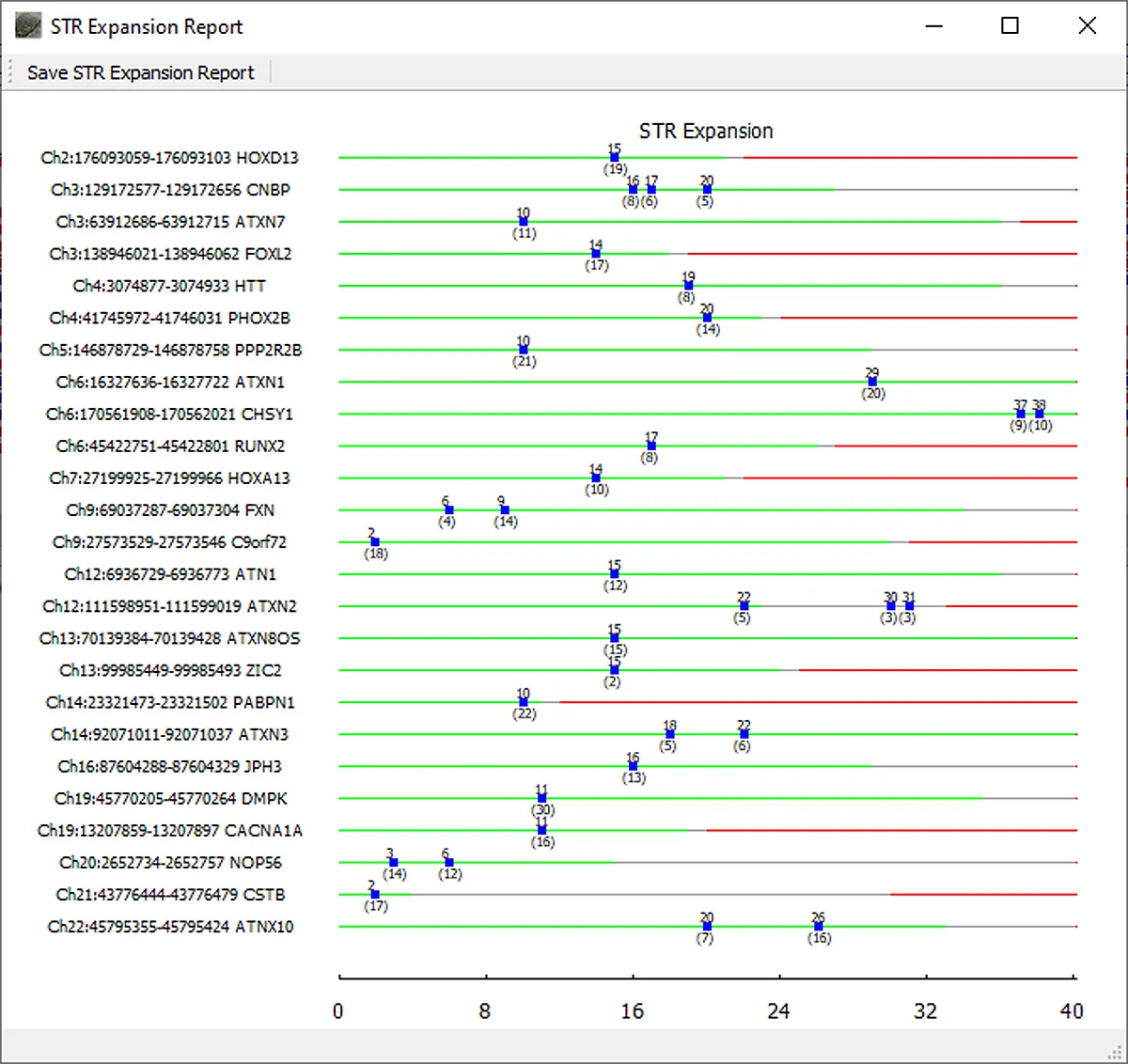
Figure 5: The NextGENeLR STR Expansion Report displays the repeat lengths detected for all STR regions of interest. Green indicates the normal STR length range, gray for premutation, and red for affected. In this case, the ATXN2 STR region shows some reads in the premutation range.
![]()
For more information on this product please contact us.



